
Weleaka. Nuyaka. Euchee. Haskell. Eufaula. Carter Seminary. These are the reeducation camps our family were forced to attend. We are the first generation not to be stolen to your federal Indian boarding "schools." This is not long ago history. It's your turn to help us carry it. #OrangeShirtDay
Recent genetic drift in the co-diversified gut bacterial symbionts of laboratory mice www.biorxiv.org/content/10.1... "Laboratory Grown Mouse strains of these ancestral symbionts have experienced accelerated accumulation of genetic load during the past ∼ 120 years of captivity"

Laboratory mice ( Mus musculus domesticus ) harbor gut bacterial strains that are distinct from those of wild mice[1][1] but whose evolutionary histories are poorly understood. Understanding the diver...
CELEBRIMBOR is now out in Bioinformatics! If you want to identify core genes that are systematically underestimated in bacterial MAG data, or you're just interested to see how we made that acronym work, go take a look: academic.oup.com/bioinformati...
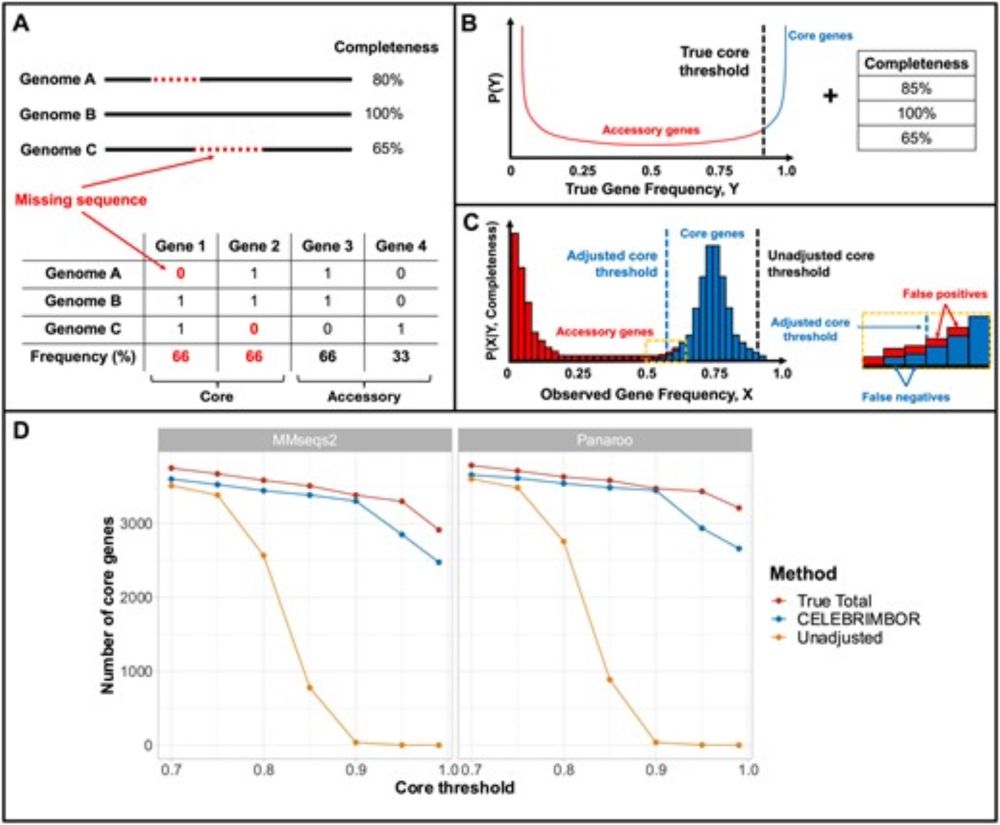
AbstractMotivation. Metagenome-Assembled Genomes (MAGs) or Single-cell Amplified Genomes (SAGs) are often incomplete, with sequences missing due to errors
The McGill freshwater ecology group is advertising for a postdoc. Come join me and six other profs! scasscsa.mcjobboard.net/jobs/165974

Thanks Mike!
Out now: genomics analysis of Pseudomonas in bronchiectasis — expanding geography & increasing 10-fold publicly-available genomes. High diversity between patients, key genes undergoing parallel evolution or diversification, differences & similarities to CF. www.journalofinfection.com/article/S016...
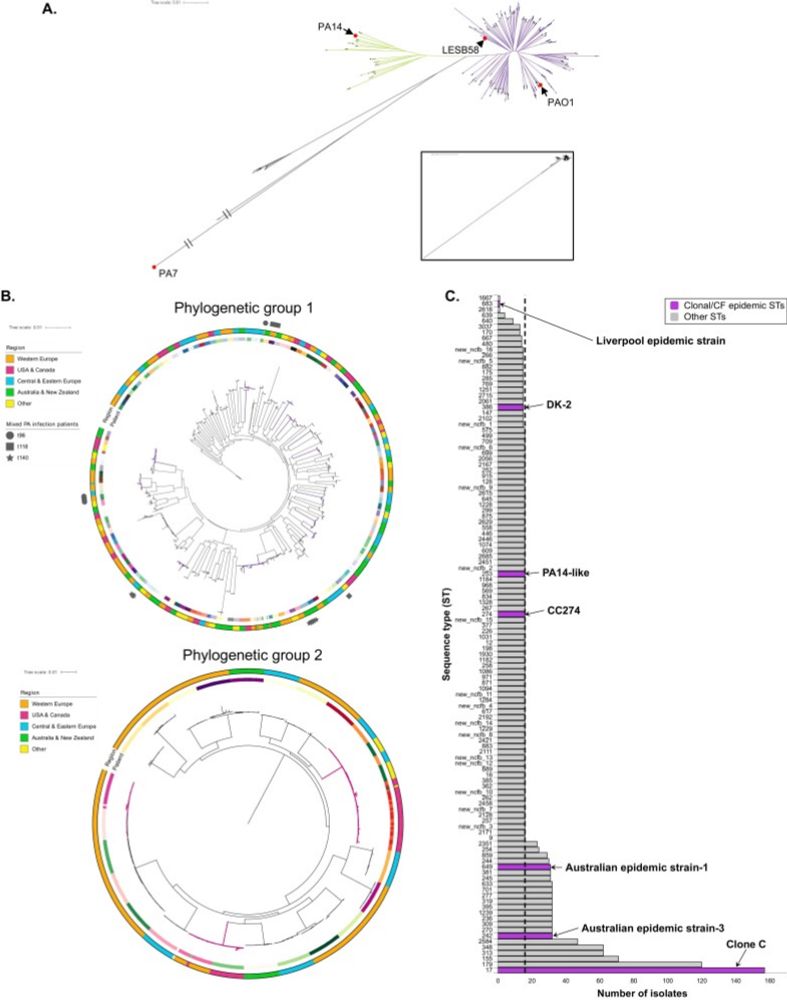
Pseudomonas aeruginosa is the most common pathogen in the bronchiectasis lung, associated with worsened outcomes. P. aeruginosa genomic studies in this context have been limited to single-country, Eur...
Alright, this thread is long enough! Congrats again to @emderrick.bsky.socialgault.mcgill.ca/en/news/deta...

Given the high levels of standing diversity in our pond species (eg pre-existing strain variation), adaptive variants could arise on different genome backgrounds. To distinguish recombination-driven gene-sweeps from homoplasy-driven soft sweeps will require other methods (sorry metagenomics). 16/n
I started off with the contrast between genome-wide and gene-specific selective sweeps. But there’s a 3rd possibility that I think is good fit to our data: soft sweeps on standing diversity. These would “look” like gene-specific sweeps because genome-wide diversity is preserved. 15/n
So we can identify targets of selection that are common across species. That’s pretty cool. A big caveat is that we can only work with species at high enough frequency in metagenomes in both control and treated ponds. Rarer species might be undergoing genome-wide sweeps and we wouldn’t know.