Projects like this can’t be completed without many others: thanks to Mark Daly for continued mentorship across the years; critical support and work from @anneotation.bsky.social@konradjk.bsky.social@gnomad-project.bsky.social. 10/10
If this work feels familiar, it is because it is building off older work from our team originally released for ExAC. I view this iteration as more of a franchise reboot instead of a sequel – we have new leads, but similar themes. 9/10


As with all gnomAD-led projects, we’ve already shared the data and code. Regions are displayed for v2 on the gnomAD browser, the code can be seen on Github (github.com/broadinstitu...), and MPC scores are available for download. 8/10
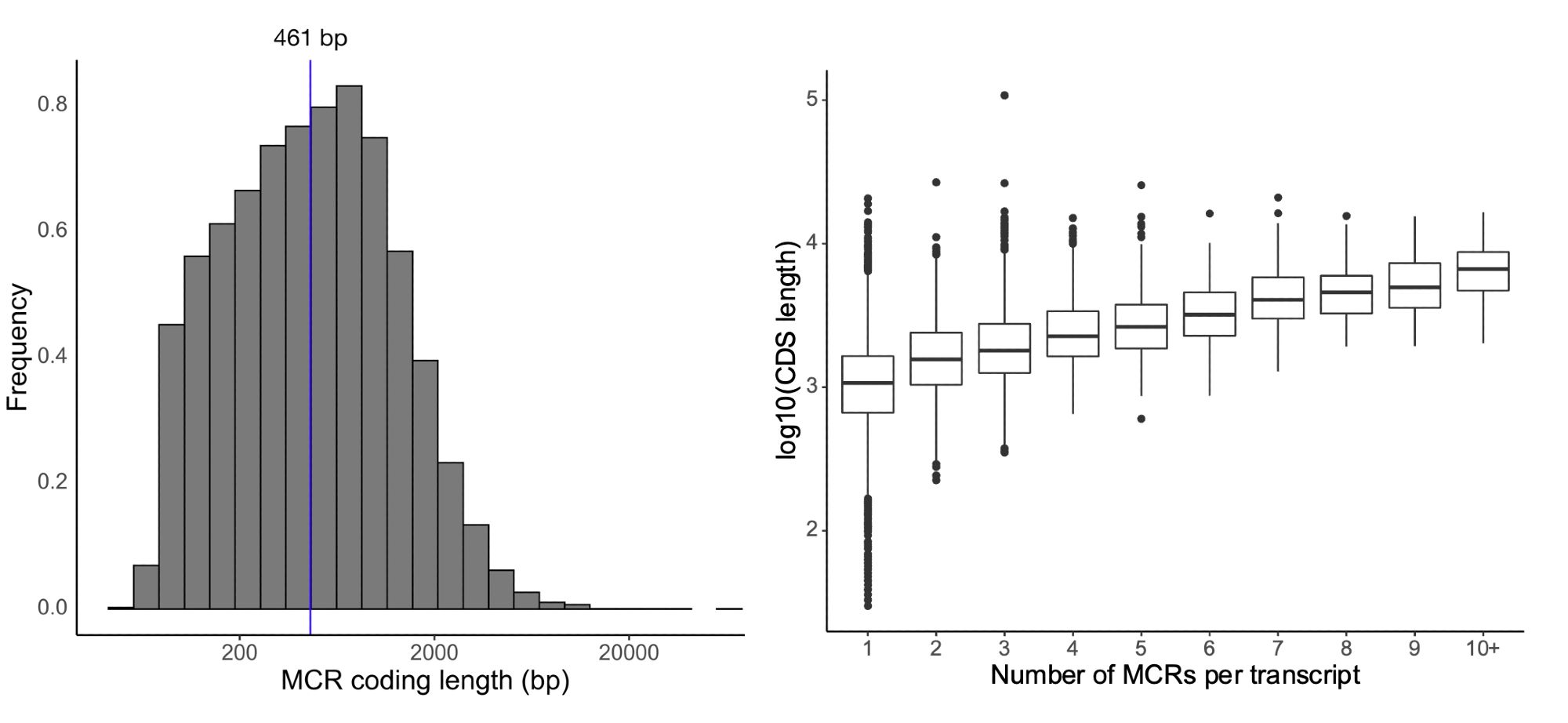
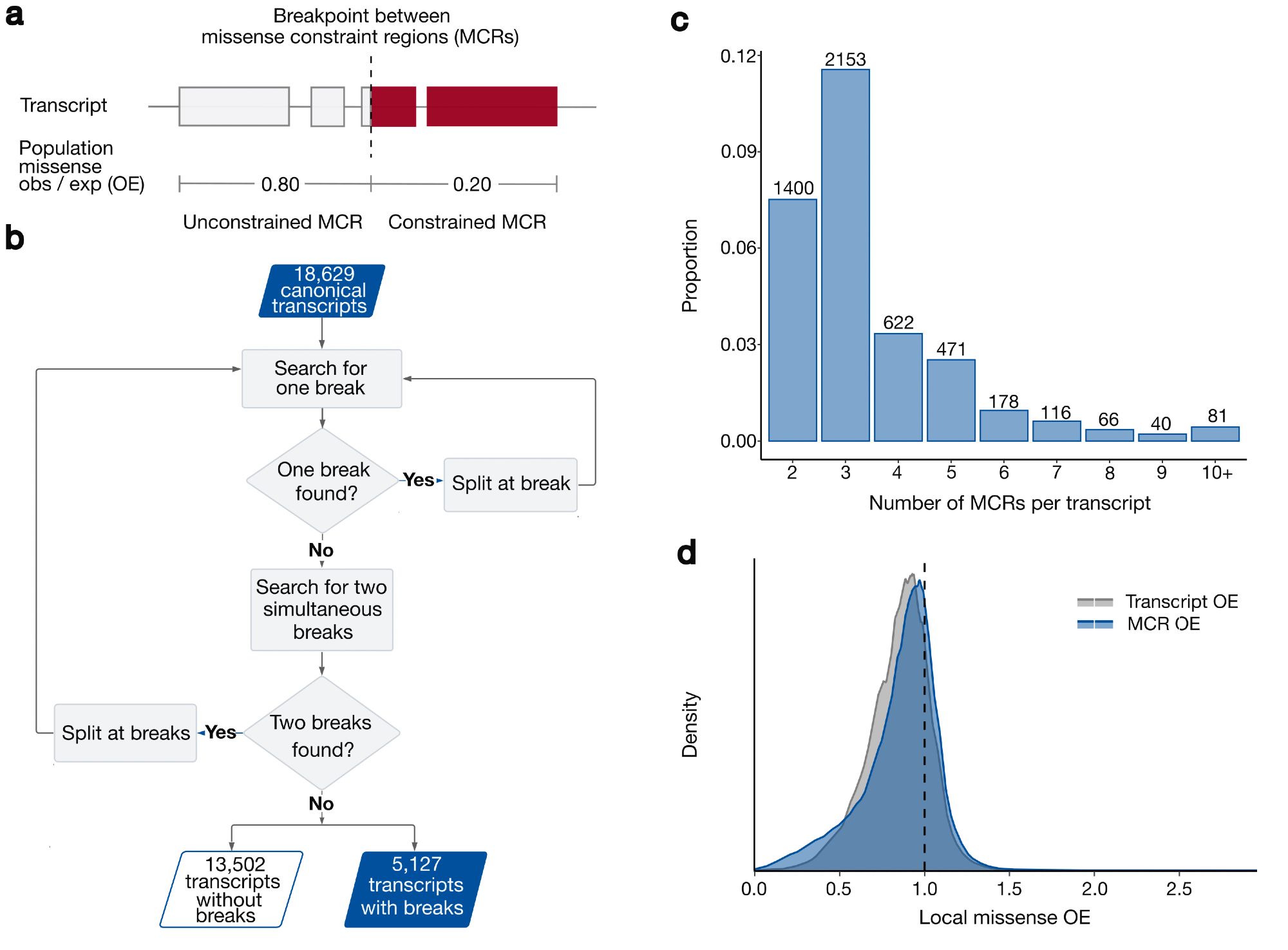
There is still much more to learn: using 125k exomes, our median region size is ~450bp and we see a relationship between transcript length and the number of regions we can identify due to statistical power. 7/10
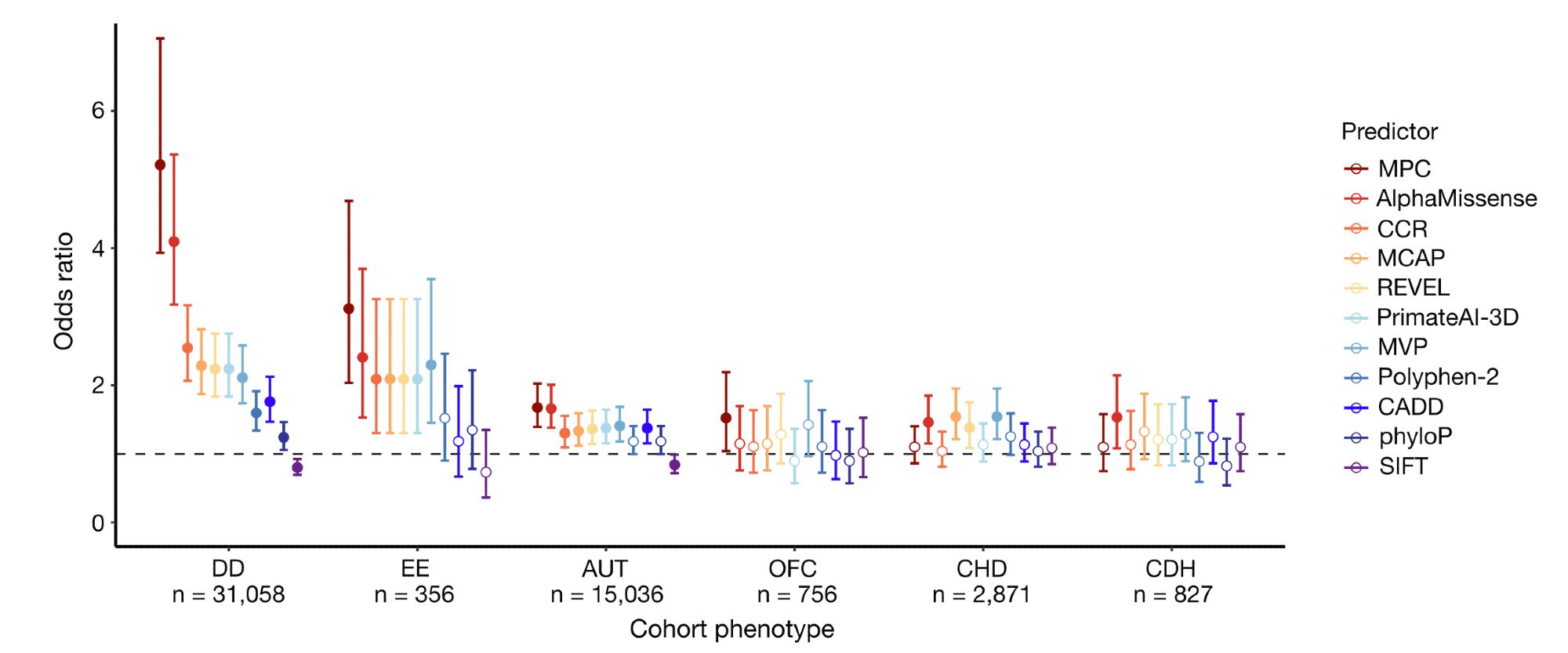
Finally, missense constraint information was incorporated into a deleteriousness metric named MPC (Missense deleteriousness Prediction by Constraint), which separates case from control de novo missense variants well with similar performance to ML models like AlphaMissense. 6/10
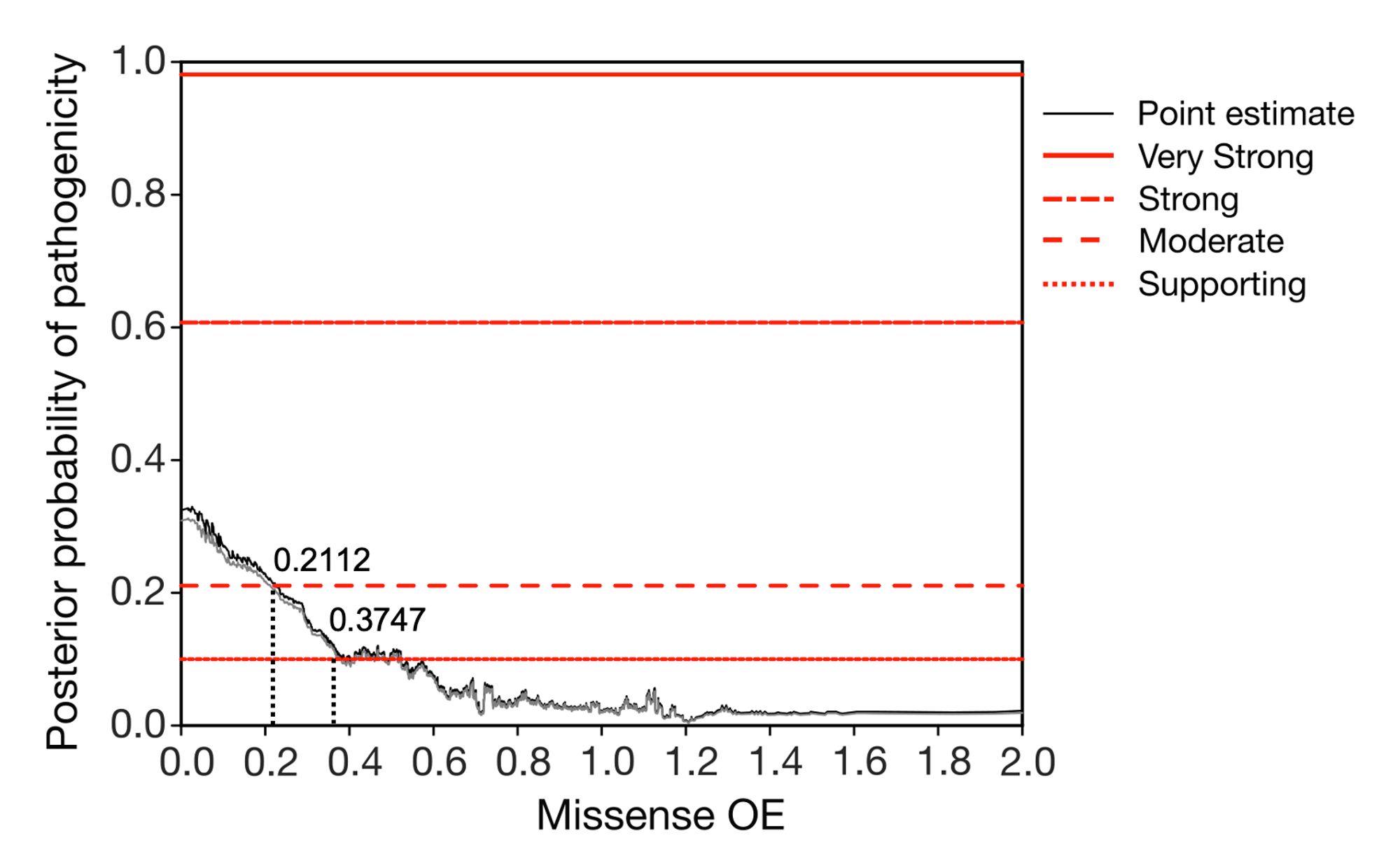
In collaboration with Predrag Radivojac and team, we demonstrated that coding bases with < 20% of their expected missense variation achieve moderate support for pathogenicity (PM1) following ACMG/AMP guidelines that can be used for clinical classification. 5/10
Missense depleted regions show an enrichment of (1) de novo missense variants in neurodevelopmental disorder cases compared to controls, (2) partitioned common variant heritability for >260 independent traits from the UK Biobank, and (3) ClinVar pathogenic (P/LP) variants. 4/10
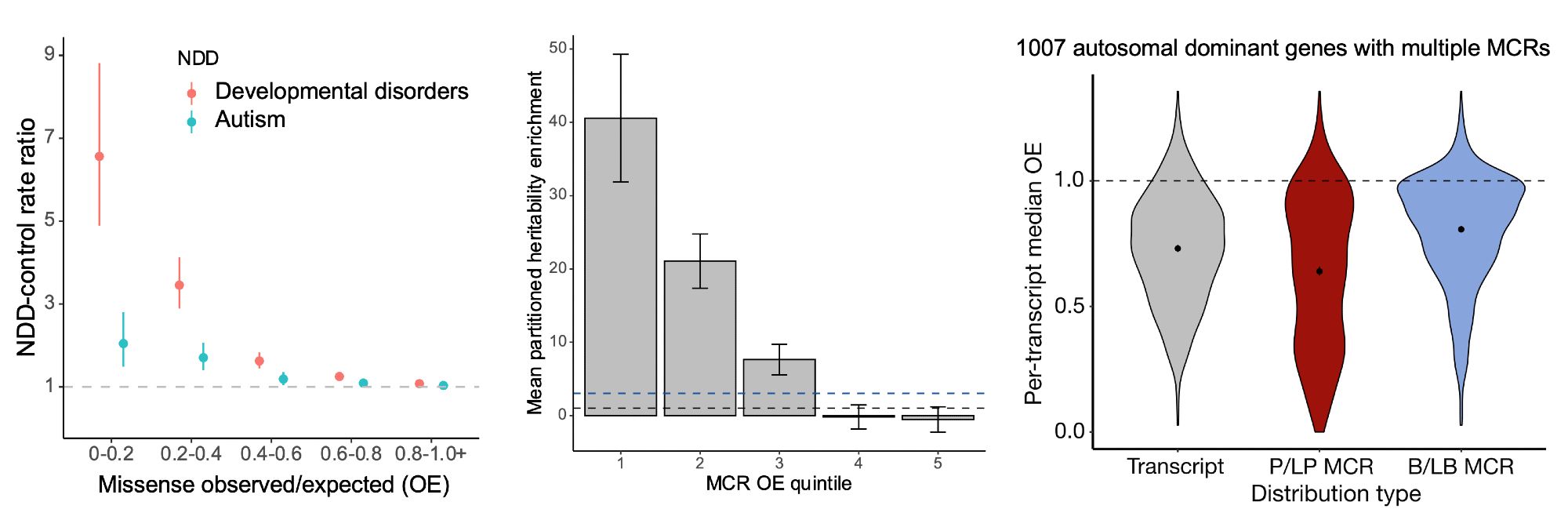
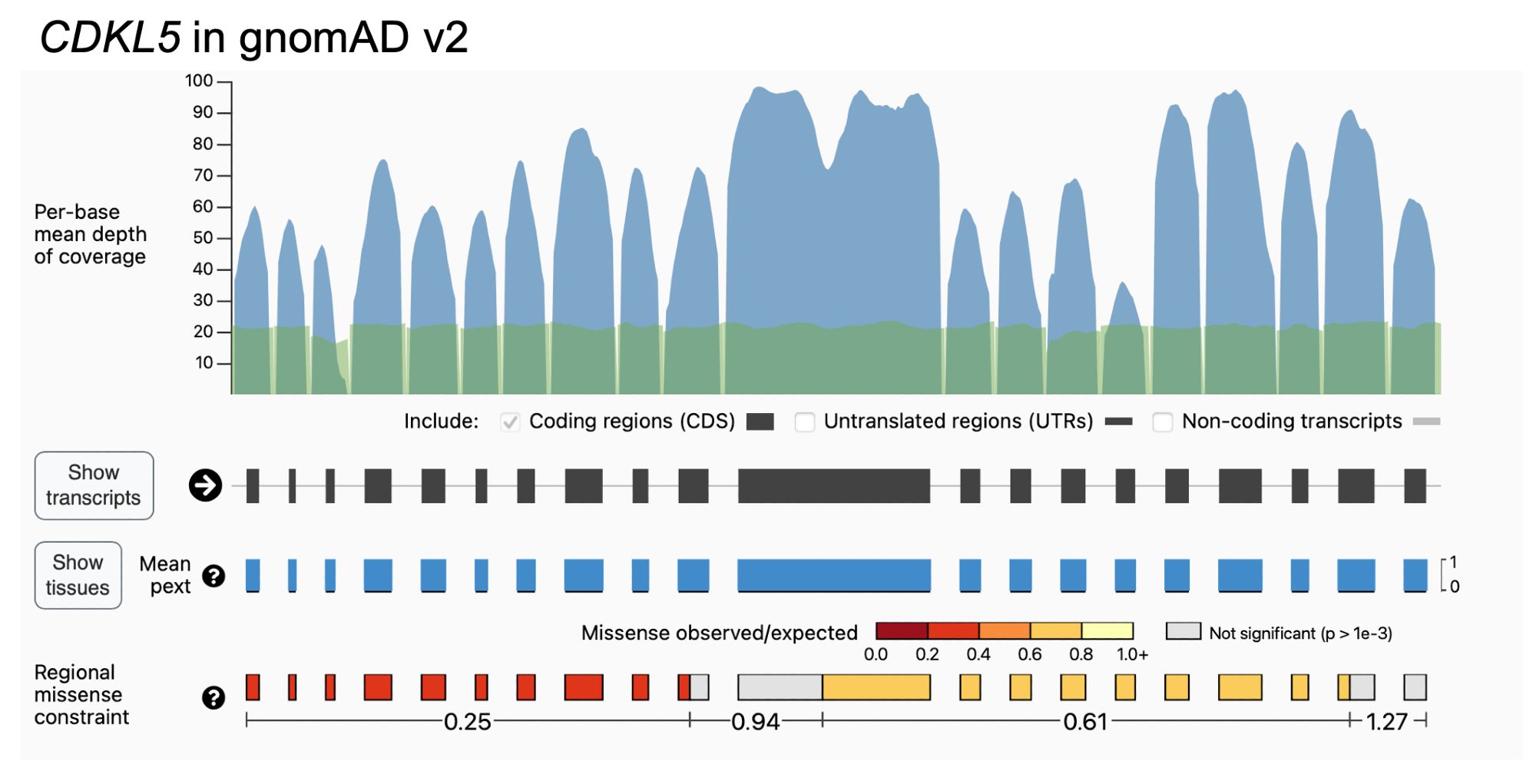
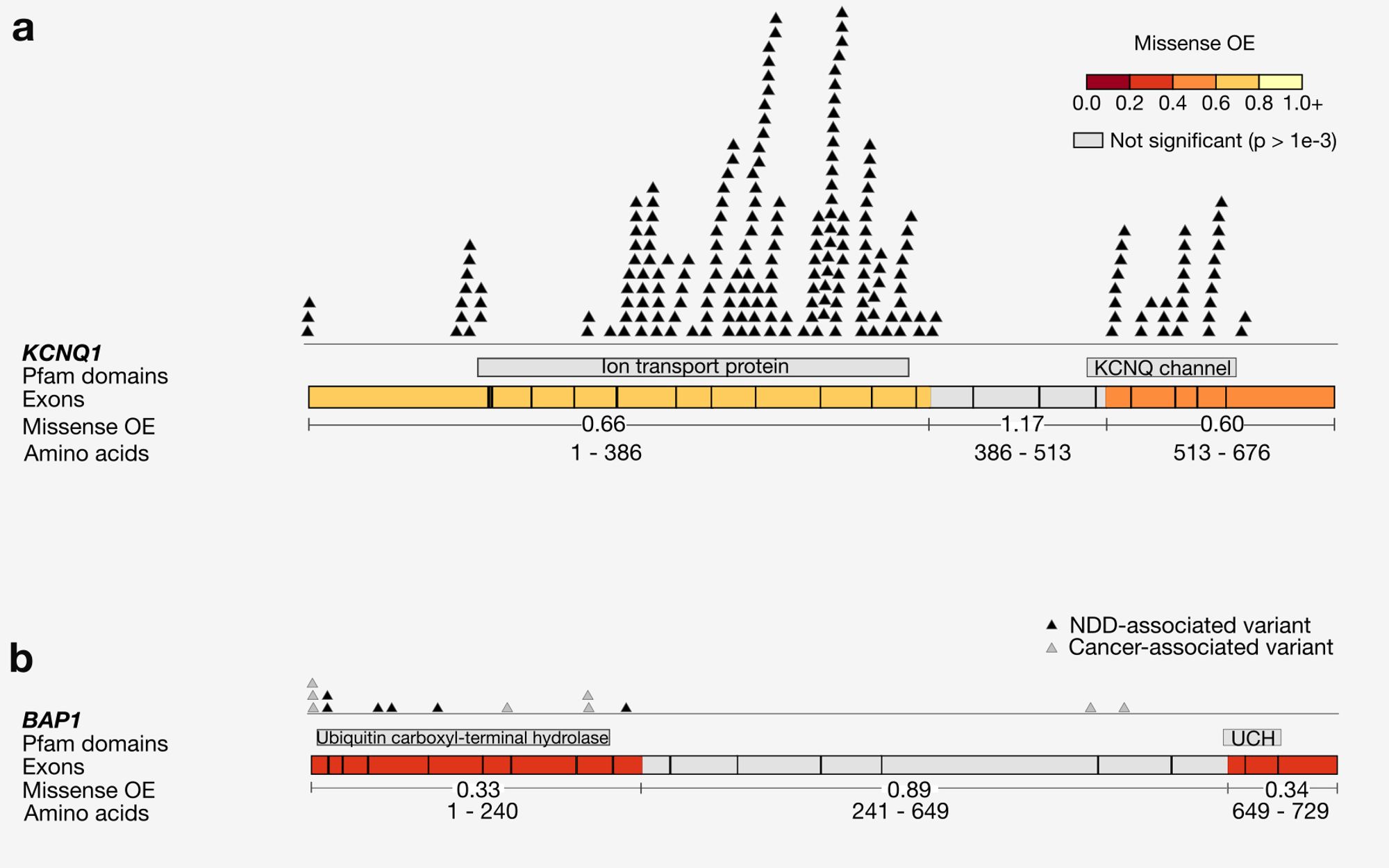
Why try to find subgenic regions that are specifically missense constrained? Splitting up genes reveals patterns of negative and neutral selection that are obscured when looking gene-wide, including highlighting regions that have a large number of known pathogenic variants. 3/10
Co-led by the fabulous Katherine Chao and Lily Wang, we used gnomAD v2 and a recursive search to identify ~28% of canonical transcripts that were split into multiple missense constraint regions (measured by variable missense depletion in gnomAD). 2/10
A freely accessible version of the paper can be found here: rdcu.be/dsVXx