An absolute game-changer for my / students' grant writing.
It's been a rough year for low-biomass microbiome research - but we've also learned a lot, and the challenges are surmountable. Here, we discuss how proactive study design and careful data analysis can help. 🖥️ 🧬 #microbiomeacademic.oup.com/jid/advance-...
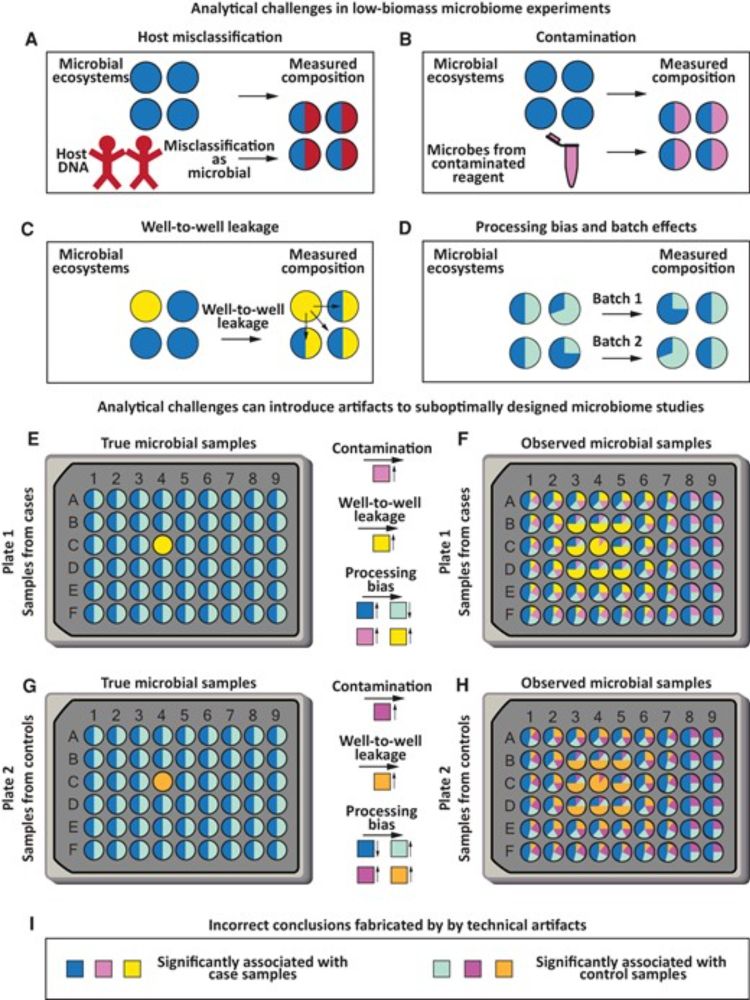
Low-biomass microbiome studies show great potential alongside major controversies. This review surveys key methodological challenges and discusses experime
New preprint! In a few words: we don't think you should use leave-one-out cross-validation (LOOCV). In a lot of words (+RebalancedCV, a LOOCV alternative): arxiv.org/abs/2406.01652 In a thread:

Cross-validation is a common method for estimating the predictive performance of machine learning models. In a data-scarce regime, where one typically wishes to maximize the number of instances...
Is finding taxa that were originally zero but have phenotype-associated values following batch correction a "major data analysis error" that invalidates downstream classifiers (in the 2020 cancer-microbiome paper or in general)? We think it's not. 📰+🧵 www.biorxiv.org/content/10.1...

bioRxiv - the preprint server for biology, operated by Cold Spring Harbor Laboratory, a research and educational institution
Check out our latest paper on how human milk oligosaccharides trigger the B. fragilis colonization program, led by Katya Buzun, in the current issue of Cell Host & Microbe! 🧪🧫🦠#MicroSky www.cell.com/cell-host-mi...
radEmu is here! ❤️🐦📈🔥 The StatDivLab's best differential abundance method yet! arxiv.org/abs/2402.05231

We consider the problem of estimating fold-changes in the expected value of a multivariate outcome that is observed subject to unknown sample-specific and category-specific perturbations. We are motivated by high-throughput sequencing studies of the abundance of microbial taxa, in which microbes are systematically over- and under-detected relative to their true abundances. Our log-linear model admits a partially identifiable estimand, and we establish full identifiability by imposing interpretable parameter constraints. To reduce bias and guarantee the existence of parameter estimates in the presence of sparse observations, we apply an asymptotically negligible and constraint-invariant penalty to our estimating function. We develop a fast coordinate descent algorithm for estimation, and an augmented Lagrangian algorithm for estimation under null hypotheses. We construct a model-robust score test, and demonstrate valid inference even for small sample sizes and violated distributional assumptions. The flexibility of the approach and comparisons to related methods are illustrated via a meta-analysis of microbial associations with colorectal cancer.
Interesting methodology + highly useful. What more can you ask for?

Come be my colleague! A broad open-rank search in areas of quantitative / computational biology - Program for Mathematical Genomics, Department of Systems Biology, Columbia University. Feel free to reach out with questions and please share. jobs.sciencecareers.org/job/653640/f...

The Program for Mathematical Genomics in the Department of Systems Biology invites candidates for faculty positions